
Population Genomics Primer I
Compiled by Julia M.I. Barth and Milan Malinsky, 21st January 2025
Table of contents
- Introduction
- Getting an overview of the data
- Calculating genome-wide population statistics
- Plotting genome-wide population statistics
1) Introduction
Background and Aim
This morning’s lecture discussed various population genetic measures and topics such as nucleotide diversity
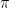 , absolute genetic distance DXY, and the fixation index FST. The aim of this activity is to get an experience of handling real population genomic data and calculating these basic statistics.
, absolute genetic distance DXY, and the fixation index FST. The aim of this activity is to get an experience of handling real population genomic data and calculating these basic statistics.
Preliminaries
We are going to work with VCF files, which summarise genetic variation across multiple individuals with respect to differences from a reference genome. If you’re interested in how a VCF is produced (mapping, variant calling, etc.) check out the “Workshop on Genomics” exercises. If required, we provide exercises to practice basic skills in UNIX and R.
- If you are new to genomics you may also benefit from this quick VCF overview exercise.
The data set
We will work with population genetic data from the cichlid fish species Astatotilapia calliptera from the crater lake, Lake Masoko in Tanzania. Two ecotypes of cichlids have evolved in this lake in the last 10,000 years or so: the deep-water, blueish, ‘benthic’ ecotype and the shallow-water, yellowish ‘littoral’ ecotype. The initial description of these ecotypes and their sympatric divergence is in Malinsky et al. (2015), while the dataset we are going to analyse comes from a more recent study: Talbi et al. (2024).
The individuals in this data set were sequenced using Illumina HiSeq paired-end sequencing to ~16x coverage, reads were mapped using BWA-mem, and variants were called jointly for 336 individuals using the GATK pipeline. In this activity, we are going to use a subset of the dataset, comprising of 69 littoral and 70 benthic individuals. For simplicity and speed, we are going to focus the analyses on chromosome 6. Other chromosomes would of course be analysed in the same way.

Learning goals
- Get an overview of a genomic dataset in a VCF format.
- Calculate key population genetic statistics from a real dataset.
- Understand basic principles and consequences of VCF filtering.
How to do this activity
- Connect to your Amazon instance via the terminal using SSH. Then go the the exercise folder:
cd ~/workshop_materials/21_popgen_primer - For R tasks switch to Rstudio in the browser (http://ec2-XX-XXX-XXX-XXX.compute-1.amazonaws.com:8787)
2) Getting an overview of the data
In population/evolutionary genomics we typically work with filtered set of biallelic SNPs. This is for a range of reasons which include: (i) they are the most common genetic variants; (ii) they can be ascertained relatively easily and accurately across individuals; (iii) we have well developed theoretical basis for working with this type of data; (iv) many analytical tools, methods, and software have been developed specifically to handle biallelic SNPs and can have difficulties with other data types. (Other types of genetic variation of course matter too; and this coming Thursday will be dedicated to larger scale structural variation and so-called pangenomes – new data structures that are great for handling large-scale structural variation.)
2.1) A first look at the VCF
We have placed a VCF file with biallelic SNPs at ~/21_popgen_primer/data/filtered_biallelic_SNPs_chr6.vcf.gz. What is in the VCF? What can it tell us about evolution of the organisms whose genetic variation is represented therein? Where do we start?
You should be able to answer at least the following questions, which can be handled with basic UNIX and with bcftools, a widely-used set of utilities is very useful for manipulating VCF files.
Show me the answer!
bcftools query -l data/filtered_biallelic_SNPs_chr6.vcf.gz
Number of individuals:
bcftools query -l data/filtered_biallelic_SNPs_chr6.vcf.gz | wc -l
Show me the answer!
bcftools view -H data/filtered_biallelic_SNPs_chr6.vcf.gz | wc -lThe
-H option to bcftools view tells it to ignore the header and only count the variants (one per line).
Show me the answer!
bcftools stats -s - data/filtered_biallelic_SNPs_chr6.vcf.gz | grep PSC > filtered_biallelic_SNPs_chr6.sampleStats
The -s - option tells bcftools to produce detailed stats for all samples (‘Per Sample Counts’) and grep PSC then searches through the bcftools output for the relevant lines.
You can find the answers by opening the filtered_biallelic_SNPs_chr6.sampleStats file in your favourite text editor. The average depth per sample is in the tenth column ([10]average depth) and counts of missing SNPs per sample are in the final column ([14]nMissing). For this number of samples, it is reasonably possible to get an overview by just looking at the raw output. However, if you have more samples, if you want to do further calculations, or for plotting, it may be beneficial to load this bcftools stats output into R.
If you go to Rstudio in your browser and navigate into the folder with the filtered_biallelic_SNPs_chr6.sampleStats file (in RStudio: setwd("/home/wpsg/workshop_materials/21_popgen_primer")), you can:
Read the data into R:
PSC <- read.table("filtered_biallelic_SNPs_chr6.sampleStats")
Plot average depth per-sample:
plot(PSC[,10],ylim=c(0,max(PSC[,10])),ylab="Average depth",xlab="Sample number")
Plot number of missing SNPs per-sample:
plot(PSC[,14],ylim=c(0,max(PSC[,14])),ylab="Missing SNPs",xlab="Sample number")
You can see that the average depth per sample is within a relatively narrow range (11.8x to 19.9x). There are no notable outliers. As for the missingness, there are two individuals that looks like outliers with >300 missing SNPs. Do we need to worry about this? One way to answer this question is to consider the number of missing SNPs in the context of how many SNPs there are in the VCF file.
Calculate missingness per-sample in percent (%):
pcMissingness <- (PSC[,14]*100)/130194
And plot:
plot(pcMissingness,ylim=c(0,5),ylab="Proportion of Missing SNPs (%)",xlab="Sample number")
We can see that the proportion of missigness per sample does not reach even 0.5%.
The above analyses would often be done as a part of quality control (QC) of a new dataset. For example, if sequencing did not work well for one individual and coverage is low, such individual would have to be removed. In this case, we are looking at a dataset that has already been QCed. Therefore, all the main measures look good and we can proceed to getting an overview of the samples and their relationships.
2.2) An overview of sample relationships
Principal Component Analysis (PCA) is a commonly used statistical method for dimensionality reduction that allows us to explore and visualise the main axes of genetic variation within the data. We are going to use the smartpca program which is described in detail in an excellent and accessible manuscript.
Because the PCA approach assumes that each SNP provides independent information, it is good practice to filter out SNPs that are in strong Linkage Disequilibrium (LD) - that is, specifically SNPs that are strongly correlated due to genetic linkage. We can do this for example with bcftools using the following command:
bcftools +prune -m 0.8 -w 50 data/filtered_biallelic_SNPs_chr6.vcf.gz -Oz -o data/filtered_biallelic_SNPs_chr6_LDfiltered.vcf.gz
This prunes SNPs with LD (r2) bigger than 0.8 in a sliding window of 50 sites along the chromosome.
The smartpca program unfortunately does not accept a VCF as an input, but it works for example with PED and MAP files. We can convert a VCF to these data formats with the commonly used genomic data analysis toolkit called plink. The following command transforms the VCF to PED/MAP while removing SNPs with minor allele frequency (MAF) of less than 5%. Here, we remove rare variants before doing the PCA to focus on the signal provided by patterns of shared common variants (the population signal) rather than signals of rare variants that reflect variants (or artifacts) specific to a few individuals.
plink2 --vcf data/filtered_biallelic_SNPs_chr6_LDfiltered.vcf.gz --maf 0.05 --export ped --out data/filtered_biallelic_SNPs_chr6
The PED and MAP files represent genotype data and variant metadata (chromosome name, ID, genetic distance (0 if unknown), position), respectively. The chromosome ID in the MAP file is the NCBI GenBank chromosome ID (LS420024.2), but smartpca expects a simple chromosome number, like chr6 or just 6. Therefore, before we can run the smartpca program, we have to change the chromosome IDs in the MAP input file. We can use the following perl one-liner to replace all occurrences of LS420024.2 by chr6 in the MAP file. Unfortunately, a lot of daily tasks in bioinformatics/genomics consist of such data conversions and troubleshooting of software compatibility issues.
perl -pi -e "s/LS420024.2/chr6/;" data/filtered_biallelic_SNPs_chr6.map
Inspect the changed file, does it look like expected? less data/filtered_biallelic_SNPs_chr6.map"
Now we are ready to run smartPCA:
smartpca -p config/PCA.par > PCA.log
The file config/PCA.par" specifies the configuration parameters for smartpca. Have a look at the file! It contains the paths to the PED/MAP input files, the names of the output files (.evec/.eval), and a specification called "numoutlieriter" which is set to 0, disabling outlier detection to include all individuals in the analysis.
After running the code above, the file filtered_biallelic_SNPs_chr6.evec should contact the eigenvectors (the directions in the data that capture the most variation, i.e., the Principal Components) and the file filtered_biallelic_SNPs_chr6.eval will contain the eigenvalues (reflecting the "amount" of variation along the eigenvectors). Before plotting the results, we need one more little format edit. This is because smartpca added the 0: prefix to each individual sample name in the output. We can remove these prefixes for example again via a perl one-liner:
perl -pi -e "s/0://" filtered_biallelic_SNPs_chr6.evec
As always, have a brief look whether the output looks like expected.
Now you are ready to switch to Rstudio and plot the results using the code in the file ~/workshop_meterials/21_popgen_primer/scripts/PCA.R.
You should be able to produce a plot that looks like the one below:
Show me the plot!

Figure: Lake Masoko ecotype PCA for chromosome 6. Individuals assigned in the field (based on the phenotype) as benthic are shown as blue rectangles and field-assigned littoral individuals are shown as yellow triangles. We see that the ecotypes separate well along the first principal component. There is one benthic individual (blue rectangle) which clusters with the littoral. This is an effect of using only chromosome 6. If you compare this PCA plot against the whole-genome PCA shown in Figure 1b of the manuscript, you will see that whole-genome data place this individual firmly as genetically 'benthic'.
Show me the answer!
filtered_biallelic_SNPs_chr6.eval file contains the eigenvalues of all the principal components of the PCA matrix. The first eigenvalue is for PC1, the second is for PC2, the third is for PC3, etc. The proportion of variance explained by each PC1 is then given by the size of the first eigenvalue divided by the sum of all the eigenvalues. In R code:eval <- scan("~/workshop_materials/21_popgen_primer/filtered_biallelic_SNPs_chr6.eval")PC1_explained <- eval[1]/sum(eval)PC2_explained <- eval[2]/sum(eval)
3) Calculating common population genetic statistics
Having established that there are two genetically distinct populations in the VCF, often the next step would be to obtain some measure of genetic variation within and between these populations. How do we calculate statistics such as π, dXY, and FST? Unsurprisingly, for a 'basic' calculation, there are various tools and approaches. But, perhaps surprisingly, the results obtained from these different approaches can vary substantially, so we have to be careful.
Our task is to calculate: (i) π in the littoral ecotype; (ii) π in the benthic ecotype; (iii) pairwise FST between the ecotypes; and pairwise dXY between the ecotypes. We are interested in genome-wide (or chromosome-wide) estimates, but also in how these statistics vary in genomic windows along the chromosome.
Key considerations:
- For FST, only variants (usually SNPs) and their allele frequencies are used in the calculation. It does not matter how many invariant sites are between the SNPs.
- In the probability activity we saw that the accuracy of FST estimates varies depending on how many SNPs are used for the estimation. Therefore, to state the obvious: genomic windows with 100 SNPs will have less variance than genomic windows with five SNPs.
- In terms of calculations, π and dXY are very similar: π measures the average number of nucleotide differences per-site (per bp) among two sequences within a population; dXY measures the average number of nucleotide differences per-site (per bp) among two sequences between populations.
- Because both π and dXY are 'per-bp' statistics, it matters how many invariant sites there are between SNPs. However, in SNP calling there are not only false positives (which we try to remove by SNP filtering), there are also false negatives. That is not all sites that are not called as a variant are actually invariant.
- For example, some sites could be called as a variant, but had insufficient mapping quality (e.g. because of being in a repetitive genomic region) or depth. So such sites would be filtered out and are now incorrectly assumed to be monomorphic (i.e. there is not a SNP in the VCF).
- Many regions of the genome are highly repetitive and it is impossible to call SNPs in these regions from short (~150 bp) Illumina reads. In a typical cichlid fish genome, around 20% to 30% of the genome is not 'callable' with short reads. Depending on the repetitiveness of the genome, it may be more or less for your organism, but it is typically a substantial proportion of animal and plant genomes.
- Such 'uncallable' regions and false negatives in SNP calling thereby lead to an underestimation of the π and dXY statistics.
- The solution to this problem is somehow keeping track during variant filtering of which sites of the genome are 'callable' and which are 'uncallable'.
In the following we are going to use and compare three methods for calculating π, dXY, and FST:
Both evogenSuite and PIXY account for false negatives in SNP calling, which is very well described in the PIXY manuscript, so do have a look if you want to understand it in more detail. On the other hand, VCFtools does not make any correction for false negatives, so it provides a good "pedagogical" comparison.
evogenSuite
To correct for false negatives, evogenSuite requires a 'callability mask' - a file that can be produced during variant filtering, defining which positions in the genome had good quality characteristics, where variants could be called confidently. We placed this callability mask file at data/accessible_genome_chr6.bed. It is in the BED file format.
Show me the answer!
awk 'BEGIN{SUM=0}{ SUM+=$3-$2 }END{print SUM}' data/accessible_genome_chr6.bed
If you wonder what this command does, or how it works, why not try and ask chatGPT?
The size of the chromosome LS420024.2 (chr6) is listed in the header of the VCF file. To read it directly, we can use:
zless -S data/filtered_biallelic_SNPs_chr6.vcf.gz
You should be able to find a line in the header that says ##contig=<ID=LS420024.2,length=41138084>.
Alternatively, you can use bcftools:
bcftools view -h data/allSites_chr6_MaskFiltered.vcf.gz | grep "##contig" | grep "LS420024.2"
The chromosome is 41,138,084 bp long. The awk command returned 30,456,555 bp. So the proportion of the chromosome that is in callable regions is 30456555/41138084 or about 74%.
Now we are ready to estimate the π, dXY, and FST statistics by running:
evogenSuite Fst --fixedW 10000 -n wpsg --accessibleGenomeBED=data/accessible_genome_chr6.bed data/filtered_biallelic_SNPs_chr6.vcf.gz config/SETS_ecotypes.txt config/PAIRS_ecotypes.txt
The --fixedW 10000 parameter tells the software to calculate the statistics in 10 kb windows. After about 1 minute run time, this 10 kb-window output will be in the file benthic_littoral_Fst_wpsg_FW10000.txt.
PIXY
Running PIXY is different from evogenSuite, in that PIXY does not take a 'callability mask' but requires as input a so-called "all-sites VCF" file. From this file PIXY makes its own assessments about which sites were callable and which were not. Such an "all-sites VCF" can be generated during variant calling, for example by using the --include-non-variant-sites flag in GATK's GenotypeGVCFs step. As the name implies, in this file, all sites — both variant and non-variant — are included.
As mentioned previously, this workshop does not deal with variant calling/filtering per se. Therefore, a ready all-sites VCF has already been prepared for you a placed at data/allSites_chr6_MaskFiltered.vcf.gz. This allows you to see what the file looks like and to get a feel for it. Perhaps the first thing you might notice: since every site is included in an 'all-sites VCF,' our dataset, consisting of 139 individuals and spanning a single chromosome of approximately 40 Mbp, results in a substantial file size of 12 GB.
To run pixy, we first need to activate the conda environment where it is installed:
conda activate pixy
Then you could run the analysis (but you probably don't want to do it now because it would take a long time; like 45 minutes at least):
pixy --stats pi dxy fst --vcf data/allSites_chr6_MaskFiltered.vcf.gz --populations config/SETS_ecotypes.txt --window_size 10000 --n_cores 2 --output_folder /home/wpsg/workshop_materials/21_popgen_primer --output_prefix PIXY
Because of the large file size and all the internal calculations needed, a PIXY run takes a long time. Therefore, we already pre-computed this for you and placed the output files in the folder ~/workshop_materials/21_popgen_primer/precomputed_outputs/. You have two options now: 1) use the pre-computed results, or 2) form teams, run the analysis in 5Mb windows, and then combine the results.
Show instructions for team-based 5Mb PIXY runs!
If you are excited about executing UNIX commands and manage to make a group of eight or nine people who like the same, then you can each run a different region and then combine the results. If you are interested in this, perhaps you can coordinate via Discord?
First let's extract a 5 Mbp region from chromosome 6 into a new VCF file and create an index for it. For example a region from 0-5,000,000:
bcftools view --threads 2 -r LS420024.2:0-5000000 data/allSites_chr6_MaskFiltered.vcf.gz -O z -o data/allSites_chr6_MaskFiltered_5mb.vcf.gz
bcftools index --threads 2 -t data/allSites_chr6_MaskFiltered_5mb.vcf.gz
Then you can run:
pixy --stats pi dxy fst --vcf data/allSites_chr6_MaskFiltered_5mb.vcf.gz --populations config/SETS_ecotypes.txt --window_size 10000 --n_cores 2 --output_folder /home/wpsg/workshop_materials/21_popgen_primer --output_prefix PIXY_5mb_0_5000000
This will take about 5 minutes, so time for some air or a quick coffee.
Afterwards you can combine these 'region files' with your team members. You probably want to do it copying them all (via scp) into a single computer.
PIXY produces three separate output files, one for π, one for dXY, and one for FST.
VCFtools
VCFtools (VCFtools website) is a "set of tools written in Perl and C++ for working with VCF files". It was developed about 15 years ago (Danecek et al., 2011) and is no longer updated or maintained. We generally don't recommend using it for anything, as most tasks it does can be achieved more efficiently using BCFtools.
Having said this, VCFtools is still regularly cited for calculations of genome-wide population statistics such as Wright’s fixation index (FST) or nucleotide diversity (π). Especially the π calculation with VCFtools is usually problematic, because the software does not account for false negatives.
To calculate π with VCFtools, we first need to split the VCF per population. To do this, we need two files with either benthic or littoral sample names. We can generate them from the config/SETS_ecotypes.txt file and then use BCFtools to produce VCF files for each population and indices for the files:
for pop in benthic littoral; do awk -v pop="$pop" '$2 == pop {print $1}' config/SETS_ecotypes.txt > config/${pop}_ecotypes.txt; done
for pop in benthic littoral; do bcftools view -S config/${pop}_ecotypes.txt data/filtered_biallelic_SNPs_chr6.vcf.gz -O z -o data/filtered_biallelic_SNPs_chr6_${pop}.vcf.gz; bcftools index -t data/filtered_biallelic_SNPs_chr6_${pop}.vcf.gz; done
Once the files are generated, briefly check if the file sizes make sense.
Then we can calculate π in windows of 10 kb. We need to do that for each population:
-
Benthic
vcftools --gzvcf data/filtered_biallelic_SNPs_chr6_benthic.vcf.gz --window-pi 10000 --out VCFtools_benthic_varOnly -
Littoral
vcftools --gzvcf data/filtered_biallelic_SNPs_chr6_littoral.vcf.gz --window-pi 10000 --out VCFtools_littoral_varOnly
We can also calculate Weir and Cockerham (1984) FST (but not dXY) with VCFtools. For FST we use the combined VCF, but we need the benthic and littoral ID files to specify the populations:
vcftools --gzvcf data/filtered_biallelic_SNPs_chr6.vcf.gz --weir-fst-pop config/benthic_ecotypes.txt --weir-fst-pop config/littoral_ecotypes.txt --fst-window-size 10000 --out VCFtools_varOnly
Finally, we could apply VCFtools to an all-sites VCF to see what results it produces in this case. Running this would again take a lot of time, so we precomputed the results and put them in: ~/workshop_materials/21_popgen_primer/precomputed_outputs/
4) Plotting the statistics
Now you can switch to Rstudio and plot the results using the code in the file ~/workshop_materials/21_popgen_primer/scripts/plot_stats.R.
As you will see, many of the results from the different analyses do not quite agree. Some even disagree by a lot. We are going to do a discussion and interpretation of these results as an interactive session.