
Where have the days gone! I’ve had to ask both Karin and Sophie to confirm what day of the week it was! Inevitably I’ve been sucked in by the Cesky Krumlov mist, several late nights enjoying fantastic genomic company, blogging in all festiveness about our good natured faculty and downloading sessions to you all…everything has started to blur. But in a good way…
Now onto Transcriptomics shall we?…
Manuel Garber
University of Massachusetts
Topic: Transcriptomics
Tonight at dinner I was trying to sum up Transcriptomics in the simplest fashion possible…
“Genes are expressed…
and then Neil (who you’ll be hearing from on Friday) chimed in…
…some more than others.”
And I think that about sums it up! Shall we all go have a drink now?
So I’m going to make you all bear with me because my background is quite minimal in terms of gene expression analysis so lets take a walk down “gene expression 101–How to run into brick walls“…for those in the class “gene expression 201–Running into brick walls and enjoying it” …skip the beginning and see the next section. For those in the class “gene expression 301–Ascending RNA-seq walls via god-like powers“, I cannot help you BUT Manuel will be waiting on the other side…
Gene Expression 101: How to run into Brick Walls
So here are some readings to get you started…alas they are not open access but if you contact Manuel I am sure he will procure these papers for you (at least the one where he is first author). If you have access then by all means dive in and have at it!
- Garber, M et al., 2011. Computational methods for transcriptome annotation and quantification using RNA-seq. Nature Methods. 8:469-477.
- Pepke, S; B Wold and A Mortazavi. 2009. Computation for ChIP-seq and RNA-seq studies. Nature Methods. 6(11 Suppl):S22-S32.
- Mortazavi, A et al., 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 5:621-628.
- Johnson, DS et al., 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316:1497-1502.
Richard Twyman writes a nice short sum up of Transcriptomics and it’s applications on the Wellcome Trust site. He defines transcriptomics as the global study of gene expression at the RNA level. So now we are not only talking about genes and their nucleotides, we are now talking about what those genes are doing, when those genes are active and to what degree those genes are active and regulated. All of this is measured through various forms of RNA (mRNA, tRNA, rRNA, ncRNA, piRNA, siRNA, microRNA or total RNA). The type of RNA you are interested in depends on what question are you asking. Richard Twyman has assisted in many research publications involving bioinformatic analysis and his recent publications can be found on the write science site.
In terms of more articles to sift through that explore transcriptomics you can try the OmicsGateway with subject: Transcriptomics through Nature Publishing, though I cannot guarantee they’ll be open access. Alternatively you can go to BMC Genomics which has a whole section on Transcriptomics where many of the articles are open access.
I’m going to go a little out of order with Manuel’s Talk…but more or less I will cover it, please see his slides for graphics, specific study notes and deeper math.
[hr]From the list of read-me’s above there are a couple different technologies/methods in place for looking at transcriptomics from a next generation sequencing bent: RNA-Seq, ChIP-Seq and today we will add on CLIP-seq. Some of the resources I am providing in this introduction are a bit outdated but are still good reviews and primers to set your mind on the right track with how to start thinking about gene expression analysis and appreciating the next generation techniques available.
Finally Manuel linked a very nice video introduction to transcriptomics that I recommend you view:
RNA-Seq: The Gene is Expressed
- In short: This technique utilizes RNA (usually total RNA, but protocols can target mRNA or other RNA or interest) that has been converted to cDNA fragments to which adaptors are attached. These fragments are then sequenced using whatever flavor of sequencing technology you prefer. Short reads can then be assembled/mapped to a reference or de novo assembled to create maps of expression a long a whole genome or genomic region of interest.
- A great introduction would be Wang, Gerstein and Snyder’s article from 2009. Again the technology has come forward since 2009, but this will give you a nice primer. Wang, Z; M Gerstein and M Snyder. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics 10:57-63 (Open Access). The article also draws comparison between sequence-based and microarray-based expression mapping.
- A nice set of slides to accompany your read through of Wang et al., can be found here as compiled by Lalit Ponnala from Cornell (simpler) and another set compiled at MIT (author unknown) (a bit more in depth). Programs are mentioned in these slides that may not necessarily be used in the workshop but feel free to explore as you see fit.
- Another slide overview of RNA-Seq that was presented 2012 and it’s still a pretty good background and was compiled by Markus Kreuz from Universitat Leipzig.
- There are quite a few applications and software packages that deal with RNA-seq analysis such as CLCbio, GenomeQuest, BioConductor and SeqMonk; however I am not going expound on these because they are not a part of the workshop; we will be using different software. But for those with interest (videos are linked instead of embedded), SeqMonk is the only one I am sure is free, the others you’ll have to take a look. Again, Manuel showed us a different pipeline but to intially set up your understanding of RNA-seq, these are nice primers for mechanistic understanding prior to jumping into the deep end of Trinity, DeSeq, RSEM and so forth…
- CLCbio Tutorial on RNA-seq analysis Part 1-pdf document
- CLCbio Tutorial via Video
- SeqMonk Website
- Bioconductor
Moving on…
ChIP-Seq: Factor Binds to the Region
In short: ChIP-seq is a way of determining how proteins interact with DNA to regulate gene expression. Genomic fragments that co-precipitate with DNA-binding proteins are sequenced. Often the DNA-binding protein of interest is a transcription factor but not always. This allows you to study all DNA fragments that are associated with your binding protein. Wikipedia actually has a nice flow of what ChIP-seq is, but essentially you cross-link protein to DNA, you then shear/fragment your DNA and add antibody to immunoprecipitate your protein. What’s left is the DNA of interest that is associated with the protein you were interested. You can then make libraries and sequence your DNA.
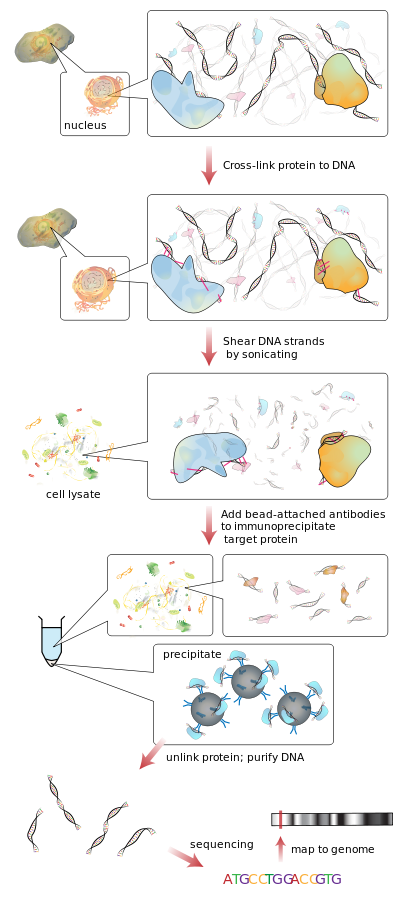
ChIP-seq therefore gives you an idea of what is being actively transcribed versus RNA-seq which will just tell you general ‘expression’, but you won’t know if that expression is ‘actively’ making protein, left over RNA from a previous bout of transcription or something else altogether.
- A good run down Q&A about ChIP-seq can be found via BMC Biology: ChIP-seq technologies and the study of gene regulation at the end of which is a wealth of articles related to ChIP-seq and it’s applications, most of which are open access. Figure 1 in the Q&A provides a schematic flow for ChIP-seq analysis.
- Nature Review Genetics also provides a nice primer article for ChIP-seq from 2009. Park, PJ. 2009. ChIP-seq: advantages and challenges of a maturing technology. Nature Reviews Genetics 10:669-680. (free)
- The ENCODE project (NCBI link to ENCODE and modENCODE) has also dabbled in quite a bit if ChIP-seq and developed some guidelines that might be of interest. Nature has an ENCODE explorer if you’d like to venture into the project from there.
- Slideshare set on ChIP-seq: Alba Jene Sanz from Biomedical Genomics Lab compiles a nice set, make sure to maximize your screen to see the small text on some of the slides. Slides 18 and 19 are nice in that they compare the different analysis tools for ChIP-seq data in table format.
As with RNA-seq here are some tools and packages for and introduction to get started on for ChIP-seq, not sure whats free and what’s not so take a look and make sure to read manuals. Some of the papers and presentations I’ve already linked also discuss tools for analysis so refer to those presentations for additional information and links.
- Bioconductor
- ChIP-seq online analysis tools site
- SeqMonk video tutorial for ChIP-seq analysis see above website in RNA-seq section for SeqMonk.
CLIP-seq: Protein Binds the RNA region
(paraphrase/adapted from Wikipedia): CLIP-Seq is short for cross-linking immunoprecipitation-high-throughput sequencing. It is a method in molecular biology used for screening for RNA sequences that interact with a particular RNA-binding protein (or another RNA). It employs UV-crosslinking between RNA and the protein, followed by immunoprecipitation with antibodies for the protein, fragmentation, high-throughput sequencing and bioinformatics. The application of CLIP-Seq methods has significantly reduced the rate of false positive predictions of miRNA binding sites and has also reduced the size of the search space for miRNA target sites.
CLIP-seq is used for: Mapping RNA/protein interactions, to find miRNA regulated transcripts, mapping translational sites, and annotating ORFs.
If you want to take a lot at some CLIP-seq databases head over to Starbase v2.0 which has a listing.
[hr]Now how about a Review? Everybody loves a good review article…
Though Pepke, Wold and Mortazavi’s paper from Nature Methods (linked above) is not freely available, BUT paywalls be damned! Slides based on the paper are available!! Huzzah and Thank You Princeton!
Once you have the sequence data then the problems turn to the computational:
Gene Expression 201: Running into Brick Walls and Enjoying It…
Read Mapping
This is similar process we’ve already learned about where you find a ‘seed’ and match it to the reference genome and ‘extend’ out from there. You can also perform assemblies de novo, but the following will focus on what you can do when you have a genome reference. Important aspects of assembly are base quality and mapping quality, if you are interested in the math equations that calculate these probabilities see Manuel’s slides. Some considerations for assembly of the data include the trade off between sensitivity, speed and memory. Assembling the data is very memory intensive, like many NGS applications you have to consider the size of your dataset in terms of what kmer length to start your seeds at for example.
- If you use a Burrows-Wheeler (BWT) based algorithm it’ll rely on a perfect match for your seed before extension. If the algorithm comes across a mismatch then it backtracks and starts all over, this is computationally expensive. As seeds get longer, then more novel algorithms are needed to match the seed in the presence of mismatches and allow for extension because longer read mapping cannot be done with BWT algorithms.
- When short read aligners come across a ‘long’ read they break it up into smaller kmers then use BWT (because now you have shorter kmers) or seed-hashing to extend the alignment.
- In terms of accounting for base quality: BWA, Stampy and Maq are all programs with algorithms that include base quality in the mapping.
In terms of RNA-seq specifically the challenge comes in mapping spliced reads where the ends occur in two different exons and cross an intron. GSNAP is a good program for dealing with spliced reads and it is a seed-hash based assembly/aligner. It also uses a seed extension method. A different program called TopHat will use an ‘exon first’ method mapping all reads that map to exons in full at the expense of your spliced read hits and it a BWT based algorithm (Bowtie2 also uses BWT).
A nice tool for visualization of assembles is the Integrative Genome Viewer (IGV) from the Broad Institute that allows you to see where the reads map and in terms of expression calculation you can load read counts to get an idea of to what extend genes/exons are expressed. Mike Zody has let me upload slide from Dr. James Robinson which I have placed on the Software IGV page on Evomics


Reconstruction
Once mapping is done, you want to reconstruct the transcript. There are several challenges to this:
- Genes exist at many different expression levels spanning several orders or magnitude.
- Reads can originate from both mature and immature mRNA and it’s difficult to tell the difference
- Reads are short and genes can have many isoforms making it difficult to tell from which isoform the read came from.
What’s an isoform? A different protein made from the same segment of mRNA that’s been alternatively spliced. So if you have a segment of Pre-mRNA, depending on how it is spliced, it can produce more than one protein or expression signature.

Scripture is a program for reconstruction of the transcripts/transcriptome that is statistically based and genome guided.
- Step 1 aligns the reads allowing for gaps that are flanked by splice sites (that we have reads for).
- Step 2 builds an oriented connectivity graph using every spliced alignment and orienting the edges according to the flanking splicing motifs.
- Step 3 then identifies segments a long the graph
- Step 4 then identifies those segments for which there is a high significance. So if you have more than one isoform it is possible to detect them as long as their significance passes the threshold defined by the investigator. This also means though that you might miss real isoforms that were just not as robustly statistically supported.
Additional considerations when designing your experiment:
- Bear in mind that different expression patterns will be observed depending on what cell type you use so if you want samples to be comparable make sure they are all treated within the same cell type otherwise the resulting RNA-seq or ChIP-seq patterns may not be comparable.
- If you have low expression levels then you will have to increase coverage to improve your ability to reconstruct those transcripts, otherwise they’ll get lost in the ‘noise’ of the experiment.
- You need to correct for multiple hypothesis testing, Bonferroni is too conservative, Family wise correction was suggested and you can see Manuel’s slides for the math.
When reconstructing expression profiles or transcripts without a reference genome one of the better programs is Trinity which uses de Bruijn graphs but a more localized approach where it locates all kmers and only extends those that are found to be most abundant. It then removes those kmers from the ‘catalog’ of possibilities and repeats the process until all extension has been done and kmers used that can be used.
Just some notes:
- If you are looking for a highly annotated genome, hopefully you have a reference to help you but you will also need a variety of techniques to achieve full annotation, RNA-seq alone will not accomplish a full annotation.
- Genome guided programs that do transcript reconstruction are Scripture as previously mentioned and Cufflinks. Scripture was designed with annotation in mind (maximum sensitivity) whereas Cufflinks was designed with quantification in mind (maximum precision). So make sure you know which you’d prefer before selecting a program. Manuel’s slides have a nice visual of how these two methods differ in terms of output as well as comparing the two programs directly in terms of CPU hours, Total memory, genes fully constructed, mean isoforms/construction etc. (Tables are at end of Post).
- Be aware that just like with other NGS assembly and alignment programs…there can be alignment artifacts that in the case of RNA/ChIP-seq can cause the formation of ‘bogus’ isoforms or loci reconstructions. Longer reads will reduce this probability.
- By using a combination of techniques you should be able to maximize your reconstructions while minimizing time/memory cost. For example using an ‘exon first’ aligner to get all the exons out of the way (at the expense of those reads that are spliced–between exons) but then going back in and aligning just those reads that didn’t align during the exon phase and reconstruction the splice reads. In addition, by going back in with the unmapped reads and mapping them to your exon reconstruction you may also pick up more reads that mapped better to the reconstruction than de novo or to the reference.
RNA-Seq can get very expensive if you are going for a full blown expression analysis.
- For full annotation you’ll need >90 million paired end reads (numbers for eukaryotic transcriptomes).
- For quantification of isoforms you’d need >30 million paired end reads and that’s of known transcriptome.
- Altnerate RNA-seq libraries such as those focusing on the 5′ or 3′ end can still yield informative results in terms of quantification and annotation but for a fraction of the cost and a fraction of the reads needed (4-8 million paired end reads)
Some of the main points to take away when thinking about expression experiments:
- Always filter your reads and get rid of adapter and barcodes as well as evaluate overall quality; trim if needed.
- If there is a reference available, use it. It will make annotation and interpretation that much easier.
- Filter our PCR duplicates
- Filter out rRNA if you can, target mRNA
- When you quantify, always normalize.
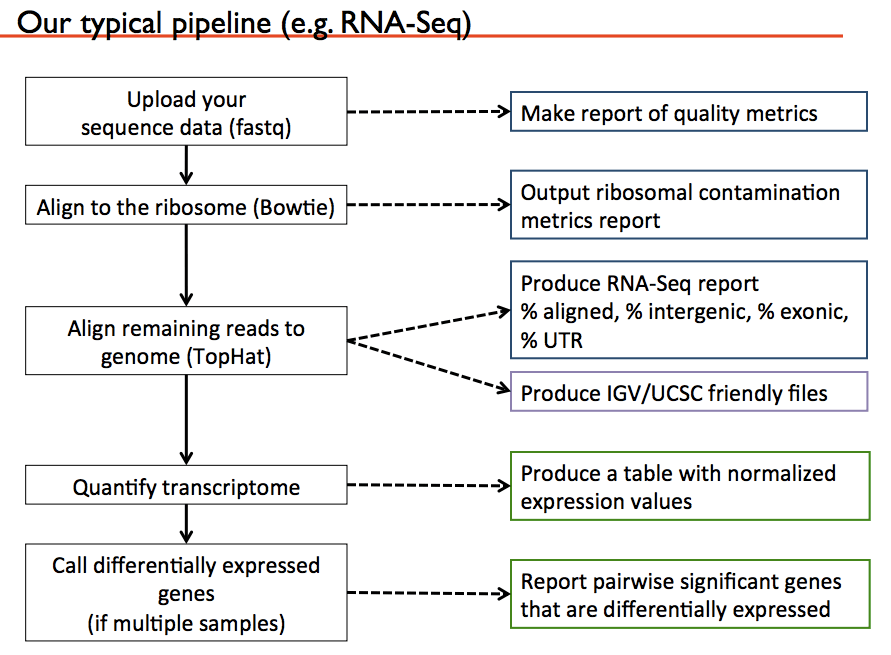
Below is a typical pipeline to consider from Manuel’s slides:
What if you don’t have a reference genome?

And yet still more considerations:
- Transcript assembly methods are the obvious choice for organisms without a reference sequence.
- Genome-guided approaches are ideal for annotating high quality genomes and expanding the catalog of expressed transcripts and comparing transcriptomes of different cell types or conditions.
- Hybrid approaches for lesser quality or transcriptomes that underwent major rearrangements, such as in cancer cell.
- More than 1000 fold variability in expression leves makes assembly a harder problem for transcriptome assembly compared with regular genome assembly.
- Genome guided methods are very sensitive to alignment artifacts.

Differences between Scripture and Cufflinks:
- Scripture was designed with annotation in mind. It reports all possible transcripts that are significantly expressed given the aligned data (Maximum sensistivity).
- Cufflinks was designed with quantification in mind. It limites reported isoforms to the minimal number that explains the data (Maximum precision).
 Quantification
Quantification
In terms of quantification, the more isoforms you have the more complicated quantifying expression becomes. In order to determine what is truly differentially expressed you should normalize your reads. Comlexity increases with more isoforms.
- For example, longer exons are going to ‘pick up’ more reads and therefore may ‘look’ more expressed but really that’s just an artifact of a longer exon to match reads too. So you have to normalize for read length.
- When you are looking across samples it is not always easy to normalize for reads because different samples might have very different inherent compositions of RNA just naturally so it’s better to use a geometric mean for that gene across all of your experiments to normalize and truly figure out what is differentially expressed.
If your goal is to tease apart isoforms then you really need paired end reads so you can use the distance between the reads to determine where isoform structures start and end. I’ve attached a figure from the slide presentation to help illustrate why paired ends make it easier to detect isoforms:
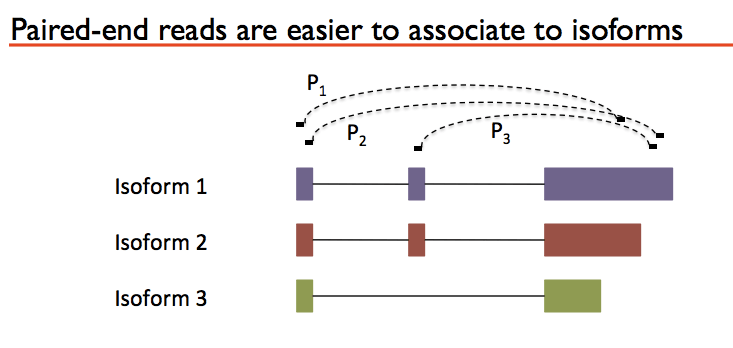
So you can see that P1 could originate from isoform 1 or 2 but not 3 because the right paired end goes beyond isoform 3. P2 and P3 you know come from Isoforma 1 because their paired ends only hit isoform one. Cufflinks is one of the programs can deconvolute transcripts (clear up isoforms).
Quantification is a complex problem in an of itself let alone looking at different expression. You can look at counts and conduct statistical tests to determine differences in expression between two conditions but bear in mind there can be bias. The larger the transcript for instance the more power you have to detect differential expression.
Be sure to Normalize!
- Normalize by total number of reads so you can compare libraries with different numbers of transcripts
- Normalize by length of transcripts
- You may have different RNA composition, that requires normalization
- Normalize your counts
- Deconvolute your isoforms by using paired reads


Gene Expression 301: Ascending RNA-seq walls via god-like powers…
As I mentioned in the introduction to this post…I really cannot help you but Manuel is waiting on the other side and I hope you have taken full advantage of his presence. Do take a look at his slides the math and equations, the statistical reasoning is all there and you probably definitely enjoyed the master class he gave us of Transcriptomic analysis no doubt. He also discusses several studies on the mouse and other different projects and experiments he and his group ran that led to all their publications and deconvolution of how to approach expression analysis.
For instance see his Vignette at the end of his slide show about lincRNA, it’s pretty cool.
Many Thanks to Manuel, gene expression has blown our minds with the possibilities and considerations…until next time.
…Dr. Mel