
Analyses of introgression with Dsuite
Milan Malinsky, Hannes Svardal, 29 January 2020
Background and Aim
Admixture between populations and hybridisation between closely related species are common and a bifurcating tree is often insufficient to capture their evolutionary history ( example ). Patterson’s D, also known as ABBA-BABA, and related statistics are commonly used to assess evidence of gene flow between populations or closely related species in genomic datasets. They are based on examining patterns of allele sharing across populations or closely related species. In this exercise, we are going to use Dsuite, a software package that implements Patterson’s D and related statistics in a way that is straightforward to use and computationally efficient. It also implements some tools and statistics which are not available in any other software, for example the f_dM introduced by Milan in this crater lake cichlid study and Hannes’ ‘f-branch’ approach from our Lake Malawi cichlid genomics paper. While exploring Dsuite, we are also going to learn or revise concepts related to application, calculation, and interpretation of the D and of related statistics.
Requirements
- Dsuite: latest version is already preinstalled on the Amazon instances
Datasets
- Simulated data: 20 populations, 5 gene flow events, 2 individuals per species
- Lake Malawi cichlids: 73 closely related species, 135 individuals
- Lake Tanganyika cichlid dataset from yesterday’s SNAPP activity
Table of contents
- Exploring a simulated dataset
- Finding adaptive introgression in Malawi cichlids
- Finding gene-flow in Tanganyika cichlids
1. Exploring a simulated dataset
We have prepared a jupiter notebook that is going to guide you through the commands and output files encountered in a typical Dsuite analysis while exploring a simulated dataset with 20 populations, 5 gene flow events and 2 individuals per species. To start working with the jupyter notebook
- Connect to your amazon cloud instance (AMI).
- Either, use guacamole. In your web browser, go to the address
- http://ec2-XXX-XXX-XXX-XXX.compute-1.amazonaws.com:8080/guacamole
where XXX-XXX-XXX-XXX is replaced by the Amazon instance IP address assigned to you. You can find that address at the web page. - username: popgen, password: same as always
- http://ec2-XXX-XXX-XXX-XXX.compute-1.amazonaws.com:8080/guacamole
- Or use ssh from your terminal
ssh [email protected](replace XXX with your Amazon instance IP address)
- Navigate into the tutorial directory:
cd ~/workshop_materials/29_Dsuite_introgression/ - Start a screen session by typing:
screen. Confirm withReturn - Start the conda virtual environment:
conda activate dsuite(we created a conda environment that contains required python packages) - Start the notebook server:
jupyter notebook - The command blocks the terminal. That is normal. Keep it running. You can get back to a functional terminal by typing
Ctrl + a, d(firstCtrl + a, thend) - In your local browser, navigate to the web address: http://c2-XXX-XXX-XXX-XXX.compute-1.amazonaws.com:8888 (replace XXX with your Amazon instance IP address, see above).
- Type the password.
- You will see the folder contents. Click on 202001_dsuite_practical_cesky_krumlov.ipynb to open the notebook and start a python kernel.
- Follow the exercises in the jupyter notebook. If you have questions please ask.
Show me how
2. Finding adaptive introgression in Malawi cichlids
This exercise is based on analysis from our manuscript published in 2018 in Nature Ecol. Evo.. The paper shows that two deep water adapted lineages of cichlids share signatures of selection and very similar haplotypes in two green-sensitive opsin genes (RH2Aβ and RH2B). The genes are located next to each other on scaffold_18. To find out whether these shared signatures are the result of convergent evolution or of adaptive introgression, we used the f_dM statistic. The f_dM is related to Patterson’s D and to the f4-ratio, but is better suited to analyses of sliding genomic windows. The calculation of this statistic is implemented in the program Dsuite Dinvestigate.
The data for this exercise are in the folder:
~/workshop_materials/29_Dsuite_introgression/MalawiCichlids/. It includes the VCF file with variants mapping to the scaffold_18 of the Malawi cichlid reference genome we used at the time. There are also two other files required to run Dinvestigate: the “SETS” file and the “test_trios” file. In this case they are called: setsFileForInvestigate.txt and test_triosForInvestigate.txt. The “test_trios” file specifies that we want to investigate the admixture signal between the Diplotaxodon genus and the deep benthic group, compared with the mbuna group. The “SETS” file specifies which individuals belong to these groups. Finally, the command to execute the analysis is:
Dsuite Dinvestigate -w 50,25 scaffold_18.vcf.gz setsFileForInvestigate.txt test_triosForInvestigate.txt
The -w 50,25 option specifies that the statistics should be averaged over windows of 50 informative SNPs, moving forward by 25 SNPs at each step. The run should take a little under 10 minutes. We suggest you have a tea/coffee break while you wait for the results ;).
The results are output by Dsuite into the file mbuna_deep_Diplotaxodon_localFstats__50_25.txt. A little R plotting function is prepared for you in the same folder. Please open Rstudio and the file plotInvestigateResults.R. Use the script to load in the file you just produced (line 3) and plot the D statistic (line 6).
Now execute line 8 of the script to plot the f_dM values. Do you see any signal near the opsin coordinates? We also plot the f_d statistic. As you can see, the top end of the plot is the same as for the f_dM, but the f_d is asymmetrical, extending far further into negative values. Finally, we zoom in at the region of the opsin genes (line 12). As you can see, the results look like a single “mountain” extending over 100kb. But there is more structure than that. Perhaps we need to reduce the window or step size to see that.
To save time, we prepared result files for runs with varying window and step sizes. They can be plotted with the same R script. Have a look at the results.
3. Finding gene-flow in Tanganyika cichlids
A Tanganyika cichlid dataset was prepared for you in ~/workshop_materials/29_Dsuite_introgression/TanganyikaCichlids. It is the same dataset that was used in yesterday’s Species-tree inference exercise. You built a “species tree”, possibly recovering the species relationships inferred by Gante et al. (2016) (shown below)?

Here we use that species tree as a basis to infer the introgression events using Dsuite. We are going to focus only on the five Neolamprologus species shown in the figure (N. marunguensis, N. gracilis, etc.). Notice that only those five species (and the Outgroup) are present in the “tree hypothesis” file GanteTree.txt, and that the usage of samples from all other species was disabled in the “SETS” file NC_031969_setsOnlyGante.txt using the xxx directive. The command to run is:
Dsuite Dtrios -t GanteTree.txt NC_031969.vcf.gz NC_031969_setsOnlyGante.txt.
This run results in only ten trio combinations of the five species, which seems manageable. Have a look at the NC_031969_setsOnlyGante__tree.txt and see if you can interpret the results. Chances are that is is still too complex. Perhaps you can try the ‘f-branch’ method:
Dsuite Fbranch GanteTree.txt NC_031969_setsOnlyGante__tree.txt > GanteTree_Fbranch.txt
You can plot the f-branch results using python code in the jupyter notebook which you used for Part 1. The results should look like this.
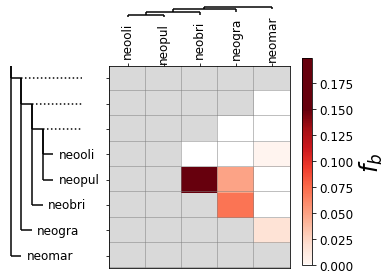{kind=link}
If you still have time, it would be interesting to see how the results look for the full set of ten species and the SNAPP tree you obtained in yesterday’s Species-tree inference exercise.
Dsuite Dtrios -t SNAPP_tree.txt NC_031969.vcf.gz NC_031969_sets.txt
Run the ‘f-branch’ command:
Dsuite Fbranch SNAPP_tree.txt NC_031969_sets__tree.txt > FullSNAPP_Fbranch.txt
You can plot the results in the jupyter notebook, or just click here.